Angew. Chem. :K+/PDA衍生的具有K-N4构型的S-区钾单原子催化剂用于有效的氧还原
在许多能源装置中,如燃料电池和金属-空气电池,氧还原反应(ORR)是必不可少的半反应。开发具有高效ORR活性和成本效益的电催化剂是这些能源技术实现商业化应用的关键。铂族金属(PGM)基材料的稀缺和昂贵性,促使研究者们致力于开发无PGM的ORR电催化剂。近年来,单原子催化剂(SACs)因其高催化活性和接近100%的原子利用率而受到了广泛关注。许多过渡金属具有丰富的d轨道电子态,合适的d带中心可以在催化反应中参与对中间体的吸附,从而提高催化活性。因此,相关领域的研究人员开发了各种过渡金属基SACs。
与常见的过渡金属元素相比,s区主族金属元素(例如K和Mg)由于"弱化学吸附",扩大了吸附质的状态,通常表现出离域s/p带,从而可能导致吸附过强(毒化吸附部位)或弱吸附(使吸附剂失活)。因而基于s区主族金属元素作为电催化剂活性中心的研究还鲜有报道。近日,我院王勤教授等人联合报道了一种具有K-N4构型的s区K SAC (K-N-C),其在碱性条件下表现出优异的ORR活性和稳定性。
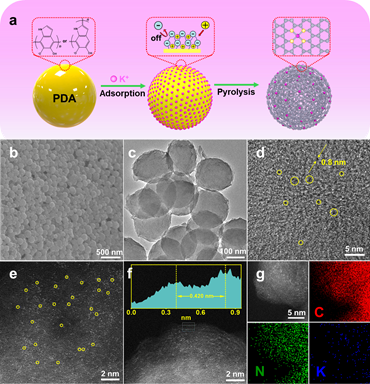
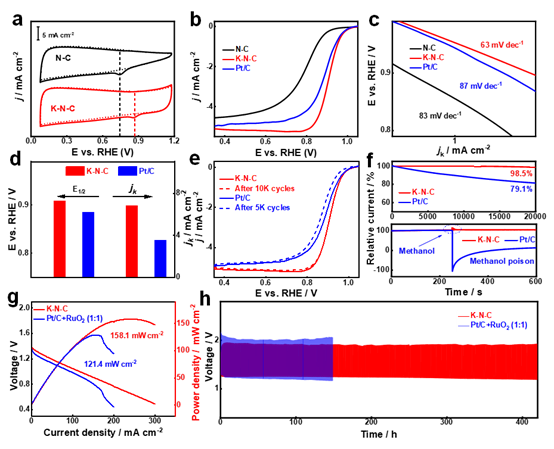
作者以K+/PDA为前驱体,煅烧后得到N原子配位的原子分散K催化剂(K-N-C)。K-N-C催化剂在ORR中表现出良好的催化活性和稳定性。半波电位(E1/2)高达0.908V。10,000次循环后,E1/2的变化可以忽略不计。此外,K-N-C提供了158.1 mW cm-2的卓越功率密度和长达420小时的锌空气电池耐久性。
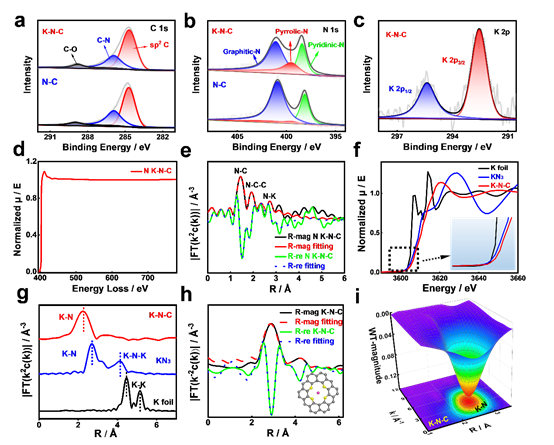
结合球差校正TEM的扩展电子能量损失精细结构(EXELFS)分析和x射线吸收精细结构(XAFS)分析确定了K-N-C中K原子的配位结构。
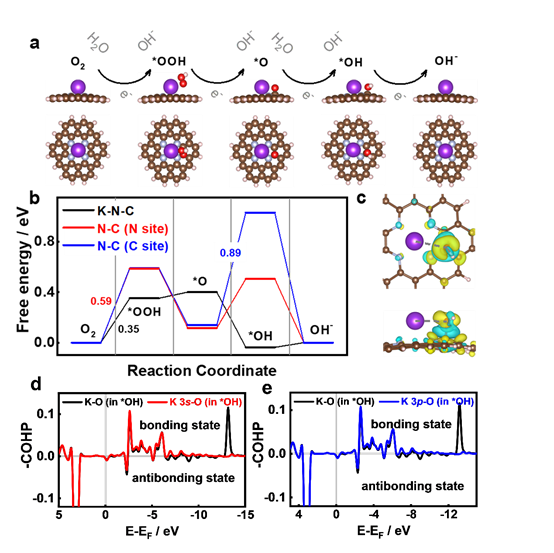
密度泛函理论(DFT)模拟表明双功能活性位点的存在是K-N-C具有高催化活性的原因。*OOH被吸附在K位点,而*O和*OH中间体被吸附在K和C位点(C-N底物中)。单位点和双位点吸附可以协同优化含氧中间体的自由能和调节速率决定步骤。 晶体轨道汉密尔顿居群(COHP)结果首次表明,与过渡金属的d轨道和碱土金属的p轨道不同,K的s轨道在中间产物的吸附中起主要作用。
该工作对s-区主族单原子催化剂的合理设计和催化机理研究具有重要的指导意义。




